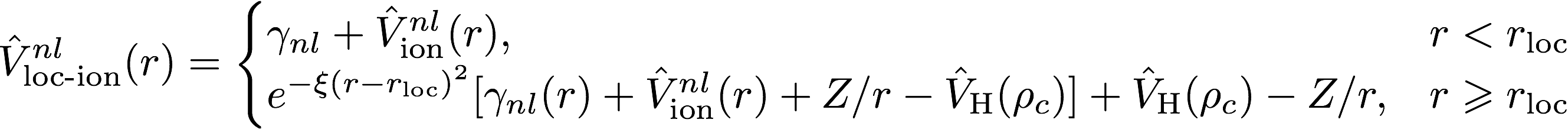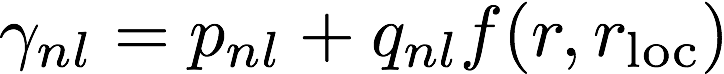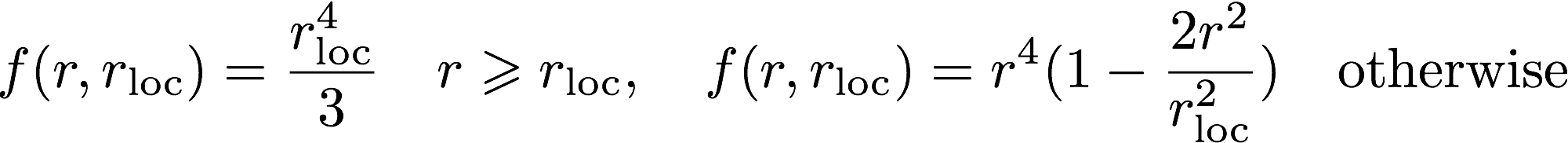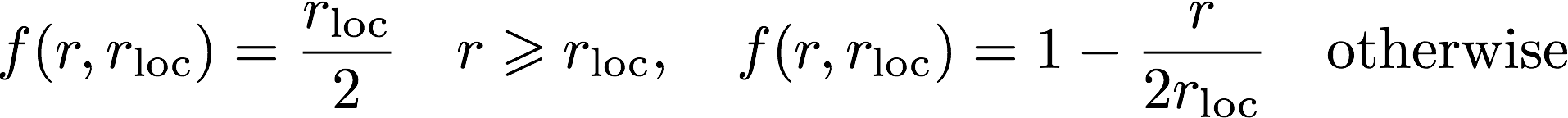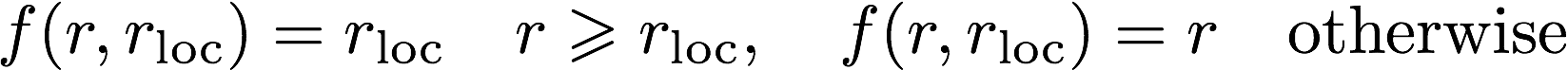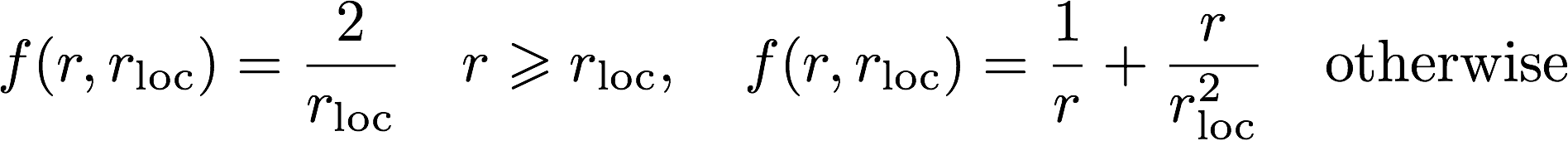The log file is the main output file for Opium. It contains all
comments, warnings, and errors. It is important to read the log file
carefully for such things. The output for the following commands will
be described below:
Welcome message:
The log file begins with a welcome message and writes out most of the
parameters of the caluclation:
++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
OPIUM Version: 1.0.2
=====================================
See http://opium.sourceforge.net for help and information
Copyright 2004 : The OPIUM project
time of execution: Thu Aug 19 16:49:42 2004
++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
Reading parameter file: <cu.param> ...
File prefix : cu
Element : Copper (Cu)
Z : 29
Number of all-electron orbitals : 8
Number of pseudo orbitals : 3
Pseudopotential is non-relativistic
Exchage-correlation functional is : Perdew-Zunger LDA
The s potential is used for the KB construction
Optimized (RRKJ) pseudopotential method will be used
nl cutoff radii q-max # bessel functions
4s 1.700 5.500 10
4p 1.900 5.500 10
3d 2.000 7.100 10
Reference Configuration: core <-/-> valence
1s2 2s2 2p6 3s2 3p6 <-/-> 4s0.75 4p0.25 3d9
Grid definitions:
a_grid=1.00e-04 b_grid=1.30e-02 # points=1131
r(1)=3.25e-05 r(np)=7.91e+01
dEmax tolerance: 1.00e-08 dVmax tolerance: 1.00e-06
Starting calculation...
|
When running non-interactively, this section is at the beginning
of every log file. In interactive mode this section is written after
every carriage return.
Back to top Back to log
All electron calculation (ae):
The first step to creating the pseudopotential is to perform the
all-electron or 'AE' calculation. Here is the all-electron output for a
copper atom:
========================================================================
Begin AE calculation
========================================================================
Performing non-relativistic AE calculation...
iter Etot Ebs Ehxc de_max dv_max
1 -3606.1898110 -2096.5570427 -1509.6327684 0.63E+00 0.47E+01
2 -3291.9228178 -1947.2651070 -1344.6577108 0.65E+00 0.10E+01
3 -3282.8597030 -1930.6798294 -1352.1798735 0.13E+00 0.18E+00
.
.
.
.
32 -3274.5405170 -1919.4887891 -1355.0517279 0.67E-07 0.66E-07
33 -3274.5405211 -1919.4887909 -1355.0517302 0.28E-07 0.13E-07
34 -3274.5405219 -1919.4887907 -1355.0517312 0.44E-08 0.26E-07
After 34 iterations...
Energy: -3274.54052195 Ebs: -1919.48879071 Ehxc: -1355.05173124
Orbital Filling Eigenvalues Norm(rc->oo)
|100> 2.000 -642.7023469980
|200> 2.000 -77.4339827449
|210> 6.000 -68.1106530315
|300> 2.000 -9.2360669866
|310> 6.000 -6.3320340017
|400> 0.750 -1.0243790023 0.8162396303
|410> 0.250 -0.5972120528 0.8703851995
|320> 9.000 -1.4633653098 0.0485860817
========================================================================
End AE calculation
========================================================================
|
The first part of the AE section shows the convergence of the
Total energy (Etot), the orbital energy (Ebs) and
the sum of the Hartree and exchange/correlation energy
(Ehxc). Also, the largest change in the eigenvalues and the
potential is printed.
The next part is the summary of the eigenvalues and partial norm
of each wavefunction (integral from the cutoff radius to infinity).
The partial norm, is only calculated for valence states.
Back to top Back to log
Pseudopotential generation (ps):
The next section is the generation of the pseudopotential or 'PS'
calculation. In this example, the optimized pseudopotential method is
used:
========================================================================
Begin PS construction
========================================================================
Optimized Pseudopotential Generation
==================
Pseudizing state : |400>
eigenvalue : -1.024379
qc : 5.500000
# bessel fxns : 10
point nearest rc : 837
rc : 1.700000
actual rc : 1.707837
Psi(rc) : 0.3826553303
Slope at rc : -0.0534146711
Curvature at rc : -0.5396774115
Del^2 Psi at rc : -0.6022298326
Log Deriv at rc : -0.1395895126
Starting KE minimization...
step # theta slope curv KEresid coeffsum
1 0.001697 -0.076480 22.534734 0.000114 0.063402
2 -0.000648 0.034498 26.600879 0.000103 0.059346
3 -0.001585 0.023718 7.481439 0.000084 0.062049
4 -0.002179 0.022481 5.158509 0.000059 0.074217
11 -0.000046 0.002442 26.290474 0.000044 0.073522
# steps: 15
Resid KE (Ry) : 0.0000430933
Norm error : 0.139E-15
Continuity error : 0.000E+00
Curvature error : -0.899E-14
Bessel wavevectors and final coefficients
1 0.4835608470 0.3552464562
2 2.6620880178 -0.2981574809
3 4.5414761759 0.0137704608
4 6.3975547999 0.0025853601
5 8.2461851245 0.0002364149
6 10.0914767940 -0.0004722742
7 11.9349823306 0.0009239812
8 13.7774199990 -0.0010797913
9 15.6191681976 0.0013383903
10 17.4604453288 -0.0011563335
Convergence term (Ry/e) : 0.0000430933
Occupation : 0.7500000000
Convergence error (mRy) : 0.0323199834
Convergence error (meV) : 0.4397392295
|
The first section displays the state being pseudized, its eigenvalue,
selected wavevector cut-off (qc), and the number of basis
functions used in the bessel expansion. Next, the grid point nearest
to the desired cut-off (pseudization) radius, the desired cut-off
radius, and then actual cut-off radius used are displayed.
The next block shows the parameters which define the soon to be
constructed pseduo-wavefunction. These are: the AE wavefunction, it's
first two derivatives, the Laplacian and the logarithmic derivative at
the cut-off radius.
The next section shows the convergence of the residual kinetic
energy minimization. The step #, step size (theta), slope and
curvature at theta=0, the residual KE and the sum of the Bessel
coefficients. After the minimization converges, the final # of steps,
residual KE and the errors in partial norm, value at rc, and curvature
at rc are all reported.
The last section shows the set of bessel wavevectors, their
coefficients and the final convergence information. The convergence
term is just the residual kinetic energy. The convergence error is
the convergence term multiplied by the occupation number.
IMPORTANT : The
convergence error should be checked carefully. This value
signifies the level of error that this state contributes to the total
energy at the minimal cut-off energy (Ecut) is qc^2 Ry. Even if you obtain
excellent transferability for your pseudoptential at, say, qc
= 3.0 (Ecut = 9.0 Ry), you still must have a small convergence error
at this qc for your results to be correct.
Once the first state is pseudized, the rest are done in turn.
------------------------------
Descreening potential
valence charge : 10.000000
core charge : 0.000000
----Solving the Schrodinger equation for all states----
State: 4s AE eigenvalue = -1.024379 PS eigenvalue = -1.024379
State: 4p AE eigenvalue = -0.597212 PS eigenvalue = -0.597212
State: 3d AE eigenvalue = -1.463365 PS eigenvalue = -1.463365
|
The next part of the 'PS' output shows total valence and core
charge. The core charge will always be zero unless a partial core
correction is used in which case the partial core charge is printed.
Next, the Schrodinger equation is solved for all valence states to
ensure that the pseudopotential does indeed yield the correct AE
eigenvalue for the reference state.
---Semilocal ghost testing---
Local state: 4s
Test state: 4p
KB energy : 12.467713 KB strength: 0.388156 KB cosine: 0.031133
el0 : -0.618203 el1 : -0.177050 eig : -0.597212
No ghosts! Ekb>0 and el0 < eig < el1
Test state: 3d
KB energy : -18.167960 KB strength: 14.284050 KB cosine: -0.786222
el0 : -0.205692 el1 : -0.085889 eig : -1.463365
No ghosts! Ekb<0 and eig < el0
No ghosts for local potential: 4s
------------------------------
Local state: 4p
Test state: 4s
KB energy : -18.202096 KB strength: 0.590062 KB cosine: -0.032417
el0 : -1.070932 el1 : -0.288800 eig : -1.024379
!GHOST! : 4s -1.024379 Should be lower than -1.070932
Test state: 3d
KB energy : -14.172017 KB strength: 11.600853 KB cosine: -0.818575
el0 : -0.202399 el1 : -0.085093 eig : -1.463365
No ghosts! Ekb<0 and eig < el0
!WARNING! Ghosts for local potential: 4p
------------------------------
Local state: 3d
.
.
.
------------------------------
========================================================================
End PS construction
========================================================================
|
The final section of the PS calculation is a loop over all valence
states to check for the existence of ghost states. This ghost testing
shows you the possible angular momentum channels that could be used as
the local potential. For instance, above we see that the 'p'
potential is not a good choice for the local potential since it will
result in ghosts.
Please see [5] for more information.
Back to top Back to log
Non-local calculation (nl):
After the semi-local pseudopotential is constructed, the
Kleinman-Bylander non-local form is tested. This step is referred to
as the NL step. The eigenvalues and partial norms for the NL section
should agree with AE section since the pseudopotential was constructed
to do this.
To do the NL step, a local potential must be defined, usually just one
of the valence potentials. Opium also has the ability to use the sum
of one or a series of step functions and a valence potential.
========================================================================
Begin NL calculation
========================================================================
Using the s potential as the local potential
iter Etot Ebs Ehxc de_max dv_max
1 -83.2901858 -14.0878751 -69.2023107 0.00E+00 0.17E-07
Converged in 1 iteration (probably reference state)
Energy: -83.29018577 Ebs: -14.08787505 Ehxc: -69.20231072
Orbital Filling Eigenvalues Norm(rc->oo)
|100> 0.750 -1.0243790023 0.8162396540
|210> 0.250 -0.5972120528 0.8703851995
|320> 9.000 -1.4633653098 0.0485860792
|
Again, notice how the NL calculation reproduces the AE results. Of course,
the total energies are different since there are no core electrons.
---Non-local ghost testing---
Local state: 1s
Test state: 2p
KB energy : 12.467713 KB strength: 0.388156 KB cosine: 0.031133
el0 : -0.618203 el1 : -0.177050 eig : -0.597212
No ghosts! Ekb>0 and el0 < eig < el1
Test state: 3d
KB energy : -18.167960 KB strength: 14.284050 KB cosine: -0.786222
el0 : -0.205692 el1 : -0.085889 eig : -1.463365
No ghosts! Ekb<0 and eig < el0
------------------------------
No ghosts present for local potential
========================================================================
End NL calculation
========================================================================
|
The last part of the NL section is another round of ghost testing.
These results should be the same as the PS ghost testing if the local
potential is just chosen from the valence. If the designed non-local
method is used (some function(s) added to a valence potential) this
can change the ghost behavior so this should be checked.
Back to top Back to log
Transferability testing: (tc)
After the ae, ps, and
nl steps are completed with small convergence errors
and no ghosts for the desired local potential, the transferability
must now be checked. Transferability measures how well a
pseudopotential performs in environments other than the reference
configuration (the configuration used in the generation step). The
all-electron and pseudo eigenvalues and partial norms are computed for
each test configuration (from the [Configs] keyblock) and
written to the log.
<<<do_tc>>>
===============Configuration 1 AE Calc===============
iter Etot Ebs Ehxc de_max dv_max
1 -3656.6376989 -2099.4527600 -1557.1849389 0.63E+00 0.56E+01
2 -3261.0263906 -1928.1127034 -1332.9136872 0.75E+00 0.14E+01
.
.
.
31 -3274.9371373 -1906.6704499 -1368.2666874 0.18E-06 0.62E-07
32 -3274.9371415 -1906.6704488 -1368.2666927 0.74E-08 0.14E-06
After 32 iterations...
Energy: -3274.93714150 Ebs: -1906.67044877 Ehxc: -1368.26669273
Orbital Filling Eigenvalues Norm(rc->oo)
|100> 2.000 -642.2116645779
|200> 2.000 -76.8977081730
|210> 6.000 -67.5794262456
|300> 2.000 -8.7475989839
|310> 6.000 -5.8528500789
|400> 0.000 -0.8700997488 0.8418328331
|410> 0.000 -0.4810316412 0.8970980498
|320> 10.000 -1.0362847356 0.0736856317
.
.
.
|
===============Configuration 1 NL: Calc ===============
Using the s potential as the local potential
iter Etot Ebs Ehxc de_max dv_max
1 -90.6881561 -14.6336531 -76.0545030 0.00E+00 0.79E+00
2 -85.5600101 -11.4740303 -74.0859799 0.22E+00 0.18E+00
.
.
.
23 -83.6927176 -10.3388674 -73.3538502 0.17E-07 0.22E-07
24 -83.6927175 -10.3388673 -73.3538501 0.63E-08 0.14E-07
After 24 iterations...
Energy: -83.69271746 Ebs: -10.33886734 Ehxc: -73.35385011
Orbital Filling Eigenvalues Norm(rc->oo)
|100> 0.000 -0.8597891449 0.8454717157
|210> 0.000 -0.4756488215 0.8994808662
|320> 10.000 -1.0338867340 0.0703706398
.
.
.
.
------------------------------------------------
================================================
|
You could compute the norm and eigenvalue differences by hand using
the log output, but it is much more convenient to generate a 'report'
file using the rpt command. The report command summarizes
key information concerning the pseudopotential.
The first part of the report file is a dump of the parameter file:
##########################################################
# Opium Report File #
##########################################################
Opium version: 1.0.2
### copy of the parameter file #######################
[Atom]
Cu
8
100 2.00 -
200 2.00 -
210 6.00 -
300 2.00 -
310 6.00
.
.
.
|
Next, the AE output is summarized:
### AE report ########################################
AE:Orbital Filling Eigenvalues[Ry] Norm
----------------------------------------------------------
100 2.000 -642.7023469980
200 2.000 -77.4339827449
210 6.000 -68.1106530315
300 2.000 -9.2360669866
310 6.000 -6.3320340017
400 0.750 -1.0243790023 0.8162396303
410 0.250 -0.5972120528 0.8703851995
320 9.000 -1.4633653098 0.0485860817
E_tot = -3274.5405219488 Ry
|
Next, the convergence error and ghost testing results are printed. The first
column is the valence state, the second column is the convergence error per
electron. Next, the error per electron is multiplied by the occupation of
the state to yield the convergence error in the reference state and is
reported in mRy as well as meV. The last column states whether a ghost was
found when this state was used as the local potential. We see that only
the s potential is a valid choice for the local potential. If the ghost
testing was inconclusive, a '?' will be printed.
### PS report ########################################
Orbital Conv. error: [mRy/e] [mRy] [meV] Ghost
--------------------------------------------------------------------------
400 0.0430933109 0.0323199832 0.4397392268 no
410 0.0172931190 0.0043232797 0.0588216796 yes
320 0.1638535915 1.4746823237 20.0642327596 yes
Tot. error = 1.5113255866 20.5627936661
|
Next, the NL test results are summarized. The ghost testing column in
this table shows whether one of the non-local potentials gives a ghost
given the choice of local potential.
### NL report ########################################
NL:Orbital Filling Eigenvalues[Ry] Norm Ghost
------------------------------------------------------------------
100 0.750 -1.0243790023 0.8162396540 no
210 0.250 -0.5972120528 0.8703851995 no
320 9.000 -1.4633653098 0.0485860792 no
E_tot = -83.2901858014 Ry
|
Finally, the transferability tests are summarized and the errors are computed and
printed.
### TC report ########################################
AE:Orbital Filling Eigenvalues[Ry] Norm
----------------------------------------------------------
100 2.000 -642.2116645779
200 2.000 -76.8977081730
210 6.000 -67.5794262456
300 2.000 -8.7475989839
310 6.000 -5.8528500789
400 0.000 -0.8700997488 0.8418328331
410 0.000 -0.4810316412 0.8970980498
320 10.000 -1.0362847356 0.0736856317
E_tot = -3274.9371415016 Ry
NL:Orbital Filling Eigenvalues[Ry] Norm Ghost
------------------------------------------------------------------
100 0.000 -0.8597891450 0.8454717157 no
210 0.000 -0.4756488216 0.8994808662 no
320 10.000 -1.0338867340 0.0703706398 no
E_tot = -83.6927174850 Ry
AE-NL:Orbital Filling Eigenvalues[mRy] Norm[1e-3]
AE-NL- --------------------------------------------------------------
AE-NL- 100 0.000 -10.3106038212 -3.6388825155
AE-NL- 210 0.000 -5.3828196377 -2.3828163585
AE-NL- 320 10.000 -2.3980016394 3.3149918622
AE-NL- total error = 18.0914250983 9.3366907362
=====================================================================
AE:Orbital Filling Eigenvalues[Ry] Norm
----------------------------------------------------------
100 2.000 -642.6711429101
.
.
.
.
=====================================================================
|
The last section is the comparison of the change in energy between
configuration 'i' and 'j' (configuration "0" is the reference) for the
AE and NL atoms. This is another quantity that can be used to measure
transferability.
Comparison of total energy differences.
DD_ij = (E_i - E_j)_all-electron - (E_i - E_j)_pseudo
AE-NL- i j DD[mRy] DD[meV]
AE-NL- ------------------------------------------
AE-NL- 0 1 -5.912131 -80.438677
AE-NL- 0 2 -0.006575 -0.089456
AE-NL- 0 3 -22.830914 -310.630563
AE-NL- 1 2 5.905556 80.349221
AE-NL- 1 3 -16.918783 -230.191886
AE-NL- 2 3 -22.824339 -310.541106
|
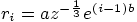 for
for 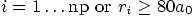 whichever comes first. This grid is used for all parts of the program
whichever comes first. This grid is used for all parts of the program